
Introduction
Artificial Intelligence (AI) is increasingly being integrated into medical devices, and in many cases the AI itself may be considered as a medical device in its own right (often referred to as AI as a medical device, or AIaMD).
As one would expect, the medical device industry is very heavily regulated. Specifically, the EU regulation of medical devices is one of the strictest and most rigid systems in the world, with manufacturers and other stakeholders needing to jump through various regulatory hoops to obtain the coveted CE mark. Medical devices must follow Regulation (EU) 2017/745, most commonly known as the EU Medical Device Regulation (MDR).
In June of 2024, Regulation (EU) 2024/1689 regulating AI products was introduced. This AI Regulation, referred to as the AI Act (AIA) lays down all the requirements for products integrating AI or machine learning (ML) depending on their risk classification. Moving forward, AIaMD and medical devices incorporating AI/ML will thus also need to consider the application of the AIA.
The EU MDR and the AIA are structured similarly as they both form part of the ‘EU new legislative framework’. Therefore, organisations already accustomed to the MDR should find the AIA to be quite familiar. In fact, there is a lot of overlap and shared definitions between these two regulations, which makes it easier for both interpretation and implementation. The goal of this article is to identify those areas that an MDR-compliant medical device will need to address to also claim compliance to the EU AI Act (AIA).
AI Act: Scope and Classification
Does my medical device fall under the EU AI Act?
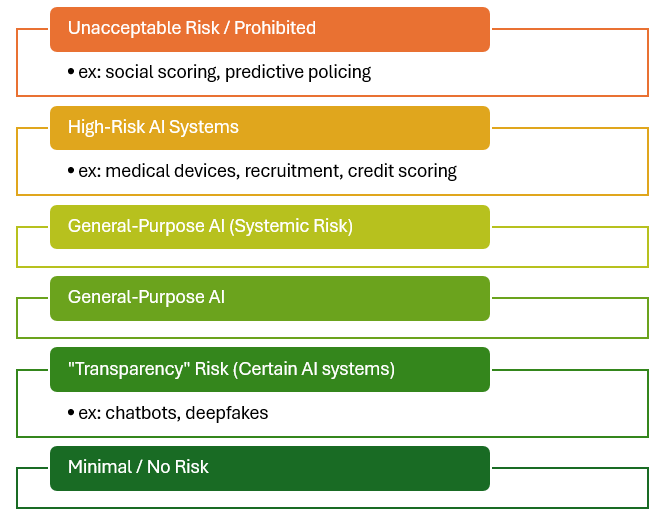
In accordance with AIA Article 2 ‘Scope’, any medical device that is or incorporates an AI system or is a General-Purpose AI (GPAI) model would fall under the scope of the AI Act.
Products falling under the scope of the AI Act are categorised into the following discrete classification levels, in reducing risk:
- Prohibited.
- High-Risk AI Systems.
- General-Purpose AI Models with Systemic Risk.
- General-Purpose AI Models.
If the AI/ML-enabled medical device does not fall under the above classifications (e.g. certain Class I AIaMD), then the provider would not be obliged to meet the AIA requirements. Nevertheless, such providers are encouraged to voluntarily adopt the AI Act measures and follow Codes of Conduct.
Prohibited
Medical devices are unlikely to fall under the ‘Prohibited’ category as Article 5 provides some exceptions to AI systems which may be used within a medical setting for a defined medical intended purpose. Whilst products which can infer emotions of a person are typically classified as prohibited, such systems intended to determine or treat a psychological indication would be allowed to be placed on the market.
High-Risk AI Systems
For a medical device to be classified as a High-Risk AI (HRAI) system, it must meet both the following criteria:
- The AI system is intended to be used as a safety component of a product, or is a product in itself, covered by the EU MDR; and
- The medical device requires Notified Body intervention under the EU MDR.
In effect, any Class IIa, IIb, or III medical device and Class B, C, D IVDs incorporating AI will be classified as an HRAI system and will need to meet the requirements of the AI Act.
General-Purpose AI Models
Medical devices considered to be GPAI models (e.g. LLM for clinical decision making), will also need to meet the AIA requirements. In cases where the GPAI model is considered to have systemic risk, there will be additional requirements to be met revolving around notification, communication, and monitoring with the relevant Competent Authorities.
Since most medical devices incorporating an AI system, or that are AI systems in themselves, require notified body intervention under Regulation (EU) 2017/745, most of these will be classified as HRAI systems. Therefore, the remainder of this article shall focus on the implications of HRAI medical devices.
Conformity Assessment
In accordance with Article 43(3) of the AIA, the HRAI medical device shall follow a conformity assessment as required by the EU MDR (typically Annex IX), however, it shall also consider HRAI system requirements set out in Section 2 of Chapter III of the AI Act. In addition to this, points 4.3, 4.4, 4.5, and 4.6 (paragraph 5) of Annex VII shall also apply.
For clarification, Chapter III contains all the requirements for HRAI systems and obligations of the various operators. Annex VII specifies the conformity assessment ‘based on an assessment of the quality management system and an assessment of the technical documentation’.
Whilst still early days, it has been heavily implied that this assessment will form part of the existing conformity assessment conducted by the manufacturer’s medical device Notified Body. With that said, the Notified Body would need to be designated for this particular type of assessment, so ensure that your existing NB has plans in place to be able to continue to serve you under the AI Regulation too.

We have a HRAI medical device. How does the EU AI Act affect us?
Provider Obligations
As a provider of an HRAI system, the AIA imposes obligations outlined in Article 16. As with similar EU legislation written under the ‘new legislative framework’, this Article does not provide much detail on how these obligations should be met, however, it does direct the provider to other sections of the regulation that must be complied with. The following is a summary of these obligations:
- Ensure the HRAI system complies with requirements in Section 2.
- Display the provider’s name, trade name, trademark, and contact address on the AI system, or where applicable, on its packaging or documentation.
- Implement a quality management system in accordance with Article 17.
- Maintain the documentation outlined in Article 18.
- Keep logs automatically generated by the high-risk AI system, as required by Article 19, when under their control.
- Ensure the HRAI undergoes the relevant conformity assessment procedure before market entry, as per Article 43.
- Prepare an EU declaration of conformity according to Article 47.
- Affix the CE marking to the AI system or its packaging/documentation, indicating compliance with Article 48.
- Fulfil the registration obligations specified in Article 49(1).
- Take corrective actions and provide required information, as stated in Article 20.
- Upon request from national authorities, demonstrate the system’s conformity with the requirements in Section 2.
- Ensure the AI system meets accessibility requirements in accordance with EU Directives 2016/2102 and 2019/882.
Quality Management System
The AIA requires that HRAI providers have an appropriate Quality Management System (QMS) that meets the requirements of Article 17. This must consider:
- A strategy for regulatory compliance.
- Design control and design verification.
- Development, quality control, and quality assurance.
- Examination, test and validation of the HRAI.
- Technical specifications and standards to be applied.
- Systems and procedures for data management.
- Risk management system.
- Post-market monitoring system.
- Vigilance.
- Communication with competent authorities and other relevant authorities.
- Systems and procedures for record-keeping.
- Resource management.
- Accountability framework detailing internal responsibilities.
If this is looking familiar to you, it’s because it should. Especially for SaMD or AIaMD products, your existing ISO 13485:2016 compliant QMS should already incorporate these points, there is nothing resoundingly different. With that said, your QMS will need to be updated to include these details of compliance with the AI Act.
For this we suggest a thorough gap assessment to be conducted against both the requirements stated in the AIA and those in ISO/IEC 42001:2023 ‘Information technology — Artificial intelligence — Management system’, currently the go-to standard for AI system QMS or ‘AI Management System’ (AIMS).
You can then integrate these requirements accordingly. It is still early days, but we expect ISO 42001 to become harmonised with the AIA in the near future.

Design and Development
After the QMS amendments have been implemented, an audit of the existing design should be undertaken to ensure that it incorporates all features and considerations required by the AI Act. The HRAI system requirements can be found throughout Section 2 of Chapter III, including:
- Design to be easily understood and transparent.
- Design for accuracy, robustness, and cybersecurity.
- Designing a system for human oversight.
- Article 50 considerations on transparency.
If needs be, retrospectively prepare documentation that shows you meet these requirements.
Technical Documentation
The technical documentation required by the AI Act shall be integrated into the existing EU MDR technical file. This avoids duplication of material over different files.
We find that the best way to do this is to perform a Gap Assessment whereby you list the requirements from the AI Act (specifically in Article 11 and Annex IV) and compare them with your existing EU MDR file. Another benefit of this approach is that you could provide assessors a clear indication of how you meet the AI Act technical documentation requirements.
There will be some deviations which can be addressed through the use of a separate document, however, it is still recommended to integrate within the existing file.
A robust EU MDR technical file will already satisfy most EU AI Act technical documentation requirements. Nevertheless, the Act focusses on Design and Development of the system.
The specifications of the contents of Instructions for Use are detailed in Article 13.
Risk Management
Risk management is already a major component of any medical device’s compliance and is a concept which is well-established throughout the industry. Article 9 of the AIA is specific to the Risk Management System for HRAIs. Reading through this, device manufacturers will immediately be reminded of the requirements specified in both EU MDR and EN ISO 14971:2019 + A11:2021. Therefore, existing risk management methodology for medical devices would be sufficient to comply with those under the AIA.
Nevertheless, we encourage HRAI medical device manufacturers to revisit their risk management and specifically make reference to the AIA within their Risk Management Plans. Since we are still in the early stages of AIA implementation, it will be critical to continually monitor any new relevant guidance or applicable standards which may be published.
Post-Market Monitoring
As many will be able to tell, Post-Market Surveillance (PMS) under the EU MDR will translate into Post-Market Monitoring (PMM) under the EU AIA. In fact, the Act requires a PMM System and subsequently a PMM Plan to be drafted up and continually conducted throughout the HRAI system’s lifetime. An official template laying down the requirements to be addressed in the PMM Plan will be published in the future. We anticipate that this will be similar to the PMS Plan requirements under the EU MDR, and may even be integrated into one Plan.
EU Authorized Representative
The manufacturer must appoint an EU Authorized Representative (AR) for the HRAI system if the provider is based outside of the EU. The obligations of an AR under the AIA are very similar to those under the EU MDR, however, these should be covered by a separate or amended mandate.
Registration Obligations
The AI Act does not require any additional registration for HRAI medical devices or their providers. Therefore, they shall solely be registered on Eudamed.
Applicable Articles of Note
AI Literacy (Article 4)
Providers and deployers of HRAI systems, especially in the medical device sector, must ensure that their staff and individuals involved in the operation and oversight of these systems possess not only a sufficient understanding of AI itself but also a strong literacy in relevant AI regulations, including the EU AI Act. This dual focus ensures that personnel can both operate the systems effectively and navigate the regulatory landscape with confidence.
Training programmes should address the technical capabilities, limitations, and risks of AI systems alongside the regulatory requirements governing their use. These programmes must be tailored to the technical knowledge, experience, and roles of staff, incorporating education on compliance obligations, data governance, risk management, transparency measures, and human oversight standards as outlined in the AI Act.

Special attention should be given to educating users about the groups or individuals on whom the systems are deployed, ensuring ethical and compliant use. Organisations can ensure safer deployment, reduce the risks of non-compliance, and build trust in AI-driven medical devices within a highly regulated environment.
AI literacy obligations begin to apply from 2 February 2025.
Blue Arrow AI Literacy Training
Data and Data Governance (Article 10)
The development of high-risk AI systems for medical devices must adhere to strict standards for training, validation, and testing datasets to ensure their quality and suitability for the intended purpose. Data governance and management practices must be implemented to oversee all aspects of dataset preparation, including design choices, data collection methods, and any pre-processing activities such as annotation, labelling, and cleaning. These practices must also address assumptions about the data’s representativeness and relevance, assess the availability and sufficiency of data, and identify potential biases that could compromise patient safety, fundamental rights, or compliance with anti-discrimination laws.
Datasets must be relevant, representative, and free from errors as far as reasonably possible, reflecting appropriate statistical properties for the system’s intended use. This includes accounting for the specific demographics, behaviours, and contexts where the medical device will operate. Any gaps or shortcomings in the datasets that could prevent compliance must be identified and addressed through appropriate measures.

Bias detection and correction are critical for ensuring equitable outcomes in HRAI medical devices. When strictly necessary, providers may process special categories of personal data for these purposes, provided robust safeguards are in place. This includes limiting re-use of the data, applying advanced privacy-preserving techniques such as pseudonymisation, and ensuring access is restricted to authorised personnel under strict confidentiality agreements. Special data must not be shared with third parties and must be deleted once bias has been corrected or when retention periods expire. Providers must document the necessity of processing such data, demonstrating why alternative methods were insufficient.
For high-risk AI systems that do not involve model training, the same stringent requirements apply exclusively to testing datasets. These measures collectively ensure the ethical and secure development of AI-driven medical devices, safeguarding patient welfare and upholding compliance with regulatory standards.
As part of the QMS, an organization must put together clear and robust Data Governance Policies that shall consider the source and type of data used for training of the AI model. It will be important to review how predicate models have been trained and retrospectively ensure the requirements of Article 10 are considered. If not, we recommend planning further studies.
Record Keeping (Article 12)
The product must have a means for logging critical events including:
- identifying situations that may result in the high-risk AI system presenting a risk within the meaning of Article 79(1) or in a substantial modification;
- facilitating the post-market monitoring referred to in Article 72; and
- monitoring the operation of high-risk AI systems referred to in Article 26(5).
Transparency and provision of information to deployers (Article 13)
Article 13 aims to establish clear requirements for providers of HRAI systems to ensure their safe, transparent, and effective deployment. It emphasizes the importance of designing systems with sufficient transparency to enable deployers to interpret outputs accurately and use them appropriately. Providers must supply comprehensive and accessible instructions detailing the system’s purpose, performance metrics, capabilities, limitations, and maintenance needs.
The information which must be provided to the deployers is shown in Article 13. This must be integrated within the IFU which must include:
Provider Information: Clearly state the identity and contact details of the provider and any authorized representative.
System Capabilities and Limitations: Explain the system’s intended purpose, performance metrics (e.g., accuracy, robustness, cybersecurity), and known circumstances that may affect performance or pose risks to health, safety, or fundamental rights. Include information on technical capabilities for explaining outputs and specific performance considerations for intended user groups.
Data Specifications: When relevant, include details about input data requirements, as well as training, validation, and testing datasets, aligned with the system’s intended purpose.
Explanation of Pre-Determined Changes: Document any changes to the system or its performance established during the initial conformity assessment.
Human Oversight Measures: Describe technical measures that enable deployers to interpret the system’s outputs, ensuring compliance with human oversight requirements in Article 14.
Resources and Maintenance Needs: Provide details about computational and hardware resources, the system’s expected lifetime, and maintenance or care requirements, including software update schedules.
Log Management Mechanisms: Describe mechanisms for collecting, storing, and interpreting system logs to meet Article 12 requirements.
Human Oversight (Article 14)
All HRAI must be designed such that they can be effectively overseen by people whilst in use. This must therefore be taken into account within the Design and Development phase of the medical device and shall include built-in measures or measures to be implemented by the deployer.
The HRAI must be provided to the deployer such that persons overseeing the system are enabled:
- To adequately understand the capacities and limitations of the system and monitor its operation. This includes detecting and addressing anomalies, dysfunctions, and unexpected performance.
- To remain aware of the tendency to over-rely on the HRAI system’s output.
- To correctly interpret the system’s output, using interpretation tools and methods, as available.
- To decide not to use the system, or disregard, override or reverse the output of the high-risk AI system;
- To intervene in the operation or interrupt the system to bring it to a halt in a safe state.
Accuracy, robustness and cybersecurity (Article 15)
Article 15 intends to establish requirements for the design and development of high-risk AI systems to ensure they maintain high levels of accuracy, robustness, and cybersecurity throughout their lifecycle. It mandates the consistent performance of these systems and encourages the development of standardised benchmarks and measurement methodologies to assess their performance.
Providers of AI medical devices must declare accuracy metrics within the IFU and implement measures to ensure resilience against errors, faults, and environmental inconsistencies, including technical redundancy and fail-safe mechanisms. For systems that continue to learn, safeguards must be in place to minimise risks from biased feedback loops.
In addition, the medical devices must be secure against unauthorised tampering or exploitation of vulnerabilities, with specific measures to address data poisoning, adversarial attacks, and other AI-specific threats. These provisions aim to uphold system integrity, safety, and reliability while mitigating potential risks.
Conclusions
Whilst the AIA introduces new requirements for certain medical devices incorporating AI/ML, many of these overlap or are very similar to those required by the EU MDR, softening the impact of compliance with the EU AI Act.
Nevertheless, providers will need to be proactive in amending their internal policies, quality management system, and technical documentation in line with the expectations of the Act. Since it is a “young” EU legislation, we should all expect constant changes in industry expectations and legislative interpretations.
How can we help?
The EU AI Act is reshaping the landscape for medical devices incorporating artificial intelligence, bringing new regulatory challenges and compliance requirements. At Blue Arrow, we specialise in guiding businesses through these complexities, ensuring seamless alignment with both the AI Act and the EU MDR.
If your medical device incorporates AI or machine learning, you’ll likely need to comply with HRAI system requirements, including data governance, transparency, human oversight, and cybersecurity provisions. Our experts can help you navigate these changes by conducting thorough gap assessments, updating your QMS, and integrating AI Act requirements into your technical documentation.
Stay ahead of evolving regulations. Partner with Blue Arrow to ensure your AI-driven medical devices meet the highest standards of safety, reliability, and compliance. Contact us today to future-proof your innovations under the AI Act.